

南湖新闻网讯(通讯员 黎学文)近日,我校资源与环境学院土壤矿物与环境团队在金属离子如何影响层状氧化铁矿物-(氧化态)绿锈的结构组成、形成演化和界面重金属吸附-氧化特性方面取得新进展,相关成果分别发表于环境地学期刊ACS Earth and Space Chemistry和Chemical geology。
该项目研究结果明确了具有不同水解速率和氧化还原特性的金属离子共存条件下,(氧化态)绿锈的形成-转化规律,结构组成特性,元素赋存形态与分布,以及界面As吸附-氧化行为和相关机制,为深入理解氧化还原交替土壤和沉积物或人造环境体系中氧化铁的形成演化规律、结构组成与特性的关联机制以及预测胶结金属元素的环境地球化学行为等方面提供了重要科学依据。
绿锈是一种层状氧化铁矿物,由带正电的Fe(II)-Fe(III)八面体片层和层间阴离子和H2O交替排列组成,广泛分布于土壤、沉积物、地下水等厌氧-缺氧环境中,显著调控着相关环境中有机-无机污染物的迁移性、毒性和环境归趋。绿锈结构不稳定,是环境中铁氧化物形成转化过程的重要中间相,当土壤氧化还原电位升高时,绿锈可通过溶解-氧化-结晶机制向针铁矿、纤铁矿等矿相转化,或通过固态氧化机制向氧化态绿锈(结构与绿锈类似,但仅含Fe(III))转化。自然环境中,各种金属离子无处不在,可通过吸附、共沉淀和同晶替代等方式进入绿锈矿相,进而影响绿锈的结构、形成转化规律和界面反应行为。然而,相关研究较少涉及。
基于此,团队选择了三种具有显著不同水解特性和氧化还原特性的二价金属阳离子(Mn2+/Ni2+/Cu2+),系统研究了不同环境条件下金属离子对绿锈形成、氧化-转化的影响及其在产物中的赋存形态与分布(图1)。金属离子共存促进了绿锈的形成与转化,且离子水解常数越大、氧化还原电位越高,促进效应越显著(即Cu2+> Ni2+> Mn2+);且少量金属离子可通过取代Fe2+的方式进入晶体结构。绿锈转化产物主要为针铁矿和纤铁矿混合相,较慢的Fe2+氧化速率和较强的Fe2+界面催化作用均更有利于针铁矿的形成,反之,有利于纤铁矿形成;三种金属离子有利于针铁矿形成的顺序为Cu2+> Ni2+> Mn2+。在转化产物中,三种金属离子有着不同的形态与分布:Mn以Mn(III)形态为主,以同晶替代方式位于结构内部且主要进入纤铁矿矿相中;Ni以Ni(II)形式均匀分布于产物晶体结构中;Cu主要以Cu(II)吸附于氧化铁表面。
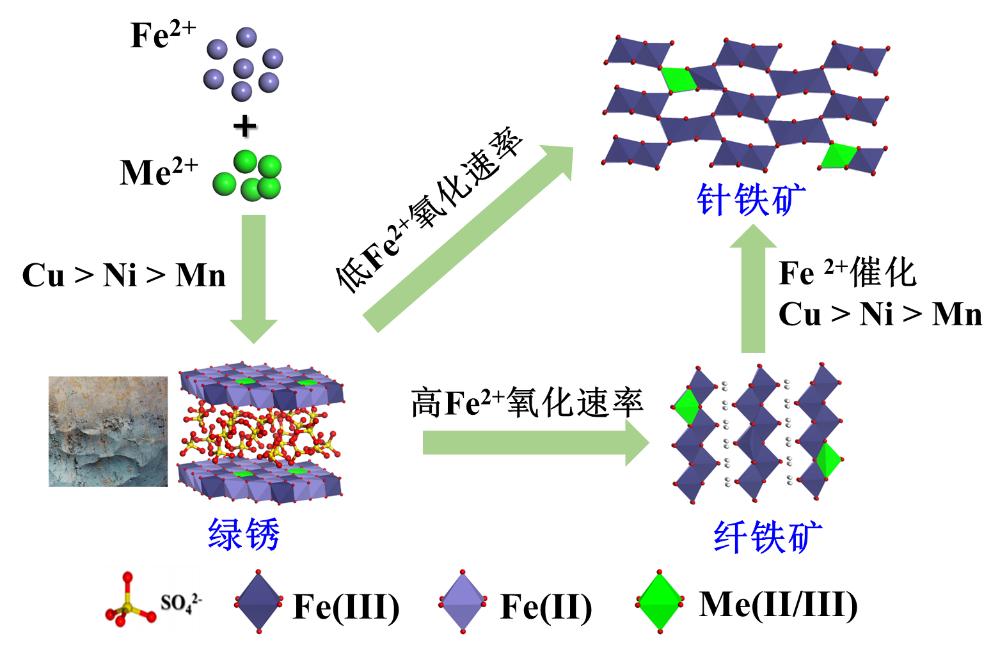
图1 在外源金属阳离子共存条件下,绿锈形成和转化示意图
此外,团队通过模拟亚表生环境条件,利用H2O2快速氧化绿锈合成系列含Mn或Al的氧化态绿锈,并通过批量实验和光谱学分析阐明了Mn或Al嵌入对氧化态绿锈的结构与组成、元素形态与分布和表面As(III)吸附-氧化的影响(图2)。Mn或Al共存有利于氧化态绿锈中富集硫酸根,大部分硫酸根位于层间,少量硫酸根和Mn或Al离子共吸附于矿物表面。Mn和Al主要以Mn(III)和Al(III)形式通过同晶替代方式进入氧化态绿锈层结构,造成c轴方向(001)面d值增加,而a-b面的d值略微减小。Al离子通过抑制绿锈结晶-生长,显著减小了氧化态绿锈的结构有序度和c轴方向铁八面体层数。与Mn嵌入相比,Al由于嵌入量更大,造成了矿物更为显著的结构变化。单位矿物质量条件下,Mn或Al嵌入促进As(III)的吸附,且进入结构的Mn(III)能够氧化As(III)。As(III)吸附涉及到表面配位和硫酸根交换两种机制,在氧化态绿锈表面形成双齿-双核内圈配位构型。这些有关绿锈研究的新认识为理解自然和人造体系中(氧化态)绿锈的矿物学特性和胶结元素的环境地球化学行为提供了重要理论基础。
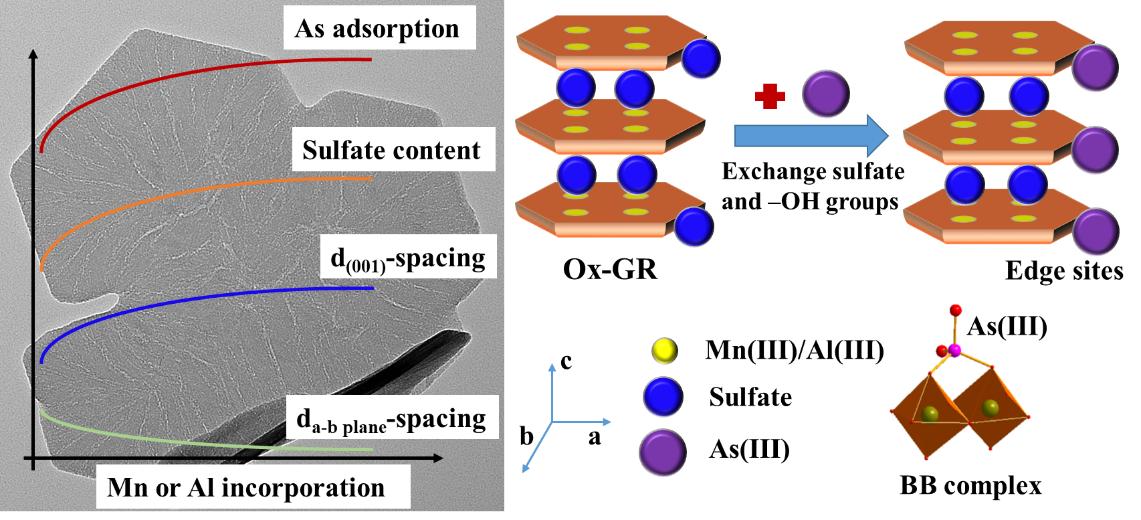
图2 Mn或Al嵌入对氧化态绿锈结构和As吸附行为的影响与相关机制
资环院王小明副教授为论文第一作者,王小明副教授和冯雄汉教授为论文通讯作者,实验数据主要由黎学文、彭晶和王兰昕等研究生获取。美国怀俄明大学朱孟强副教授和法国格勒诺布尔阿尔卑斯大学Bruno Lanson教授等参与指导了部分研究工作。本研究得到国家自然科学基金、国家重点研发计划子课题和中央高校基础研究基金的资助。
审核人:王小明
【英文摘要】
Green rusts (GRs), which are important intermediate phases during Fe2+ oxidation, are commonly associated with various metal cations during their crystallization in soils and sediments, but the effects of these foreign metal cations on the formation of GRs and on their subsequent transformation to Fe (hydr)oxides remain unclear. In the present study, the effects of Mn2+, Ni2+, and Cu2+ on the evolution processes of hydrosulfate green rust (GR2) are documented under various conditions and the mechanisms leading to cation incorporation in the reaction products are determined. The rates of GR2 formation and of its transformation to Fe (hydr)oxides both decrease in the order of Cu2+ > Ni2+ > Mn2+, and increase with increasing metal cation concentration. During GR2 crystallization, a small fraction of foreign metal cations is structurally incorporated in GR2 by replacing Fe2+, and their amount in the mineral follows the order of Cu2+ > Ni2+ > Mn2+. Under all conditions, the final reaction products are a mixture of lepidocrocite and goethite; a slow oxidation rate of mineral Fe2+ and a strong catalytic effects of surface Fe2+ both facilitate the goethite formation from GR2, reversely, favorable to lepidocrocite formation. Additionally, the three cations possess different speciation and distribution in lepidocrocite and goethite: Mn exists mainly as Mn(III) and small Mn(II)-Mn(III) molecular clusters and occurs mainly in the mineral interior by isomorphic substitution or coated by the Fe (hydr)oxides crystals; Ni is present as Ni(II) and uniformly distributed in the newly formed minerals by either isomorphic substitution or surface adsorption; finally, Cu is mainly sorbed at the mineral surface as Cu(II) with minor Cu(I). These cations may thus be structurally incorporated in Fe oxides in the order of Mn(III) > Ni(II) > Cu(II). These new insights into the interaction between GR2 and trace metal cations improve our understanding of Fe oxide crystallization processes and of the environmental geochemical behavior of associated metal cations in redox alternating soils and sediments.
【原文链接】
https://pubs.acs.org/doi/10.1021/acsearthspacechem.8b00187
【英文摘要】
The trace cations could incorporate into the structure of oxidized green rust (Ox-GR), a layered reactive Fe(III) oxyhydroxide formed from rapid oxidation of GR, but the effects of cation incorporation on the mineralogical properties and surface reactivity of Ox-GR remain unknown. Here, we synthesized Mn- or Al incorporated Ox-GR by oxidation of sulfate-bearing GR and determined their structure, elemental composition and distribution, and As(III) adsorption using macroscopic batch experiments and spectroscopic analyses. The presence of Mn or Al favored sulfate accumulation in Ox-GR, with some sulfate being homogeneously distributed in the interlayer, others adsorption on the mineral edge sites. Majority of Mn and Al entered the layer structure of Ox-GR as Mn(III) and Al(III) through isomorphous substitution, leading to the increased d spacing of (001) plane but slightly decreased d spacing of a-b planes. The Al incorporation remarkably reduced the structural ordering degree and Fe octahedral layers of Ox-GR through inhibiting the crystal-growth of GR. Compared with the Mn incorporation, the Al incorporation led to a more pronounced structural variation of Ox-GR, ascribed to its higher isomorphic substitution amount. The incorporation of Mn or Al both promoted As(III) adsorption per mineral mass, predominantly due to the increase of sulfate content and/or specific surface area, and the incorporated Mn(III) could oxidize As(III). As(III) adsorption on Ox-GR involved both surface sulfate and >Fe-OH/OH2 groups exchange, forming a bidentate binuclear inner sphere surface complexation. These new insights into the structure and reactivity of Ox-GR are essential to understanding environmental behavior of GR and its derivative Ox-GR in artificial and environmental settings.
【原文链接】
https://www.sciencedirect.com/science/article/pii/S0009254122004181